
Breakends, structural variants and CNV detection
Copy number variants (CNVs) are the subject of extensive research. They are common features of the human genome that play an important role in evolution, contribute to population diversity, development of certain diseases, and influence host–microbiome interactions. CNV analysis has found application in the molecular diagnosis of many diseases and in non-invasive prenatal care; still, its full potential lies ahead in time. CNVs are expected to have a significant impact on screening, diagnosis, prognosis, and monitoring of several disorders, including cancer and cardiovascular disease (Carey-Smith et al. 2024, DOI: 10.3390/ijms25136815 , Caputo et al. 2025, DOI: 10.3390/jcdd12070258).
While it is clear that CNVs play a significant role in the modification of the function of the genome, CNV calling has been challenging for clinical diagnostic laboratories. This was also shown in two pilot external quality assessment tests run by EMQN and GenQA together with Euformatics in 2022 and 2024, and presented first time at ESHG in 2023 (Gutowska-Ding et al. 2023, DOI: 10.1038/s41431-023-01482-x). Challenges of different kinds were identified.
On a high level, a great diversity of CNV reporting was seen for one and the same sample (Figure 1 below). Indeed, setting up the CNV caller pipeline is not mastered everywhere, leading for example to some unexpected results, such as omitting and rejecting shorter CNVs at specific thresholds. In the oppisite direction, some used pipelines also reported CNVs insertions or deletions shorter than 10 base pairs. Such variants, by convention, belong to the category of Indels (in general when shorter than 50bp) and can be called by simpler pipelines used for SNVs and Indels.
On a detailed level, break-end discovery, as a step in CNV detection, for the same sample, varied significantly with up to 1500bp for exon-based panels, while WGS-based (whole genome sequencing) breakend calling, quite expectedly, could get down to more precise position across different callers. Breakends can also for biological reasons show high variability with many unique breakpoints but also recurrent hotspots with clustering of breakends around “unstable” genomic loci is. Persistency of breakend detection, beyond technical issues of detection, is affected by genomic architecture such as chromatin structure, sequence homology, repeat content, replication timing, fragility, and of course selective pressures.
Getting better with CNV detection
In a nutshell, good detection requires a homogenous sequence signal, in other words regular, maximally uniform read depth and per base
quality.
The difference between panel sequencing, including WES on one hand, and WGS on the other is related to two challenges. Uniformity of the
coverage is essential for CNV detection. Segmented sequence information, where intron sequence is mostly lacking, makes it, if not
impossible, then at least more difficult to recognise the position of the breakends. Moreover, in segmented, mostly exon-sequencing, the
detection signal for a capture is not even over the exons, diminishing read depth near the segment ends. In theory, therefore, calling CNVs
on typical WES data can, at best, get down to a per exon breakend approximation.
For WGS data, an uneven signal landscape, also including introns, just looks like, well, capture and sequencing errors. Consequently also here
breakend identification is affected by genome architecture. Noise or real signal, that will be the question for any pipeline. For practical reasons
also price of WGS sequencing and data management of complete genome data can be an issue for clinical diagnostic laboratories and WES still maintains some advantages.
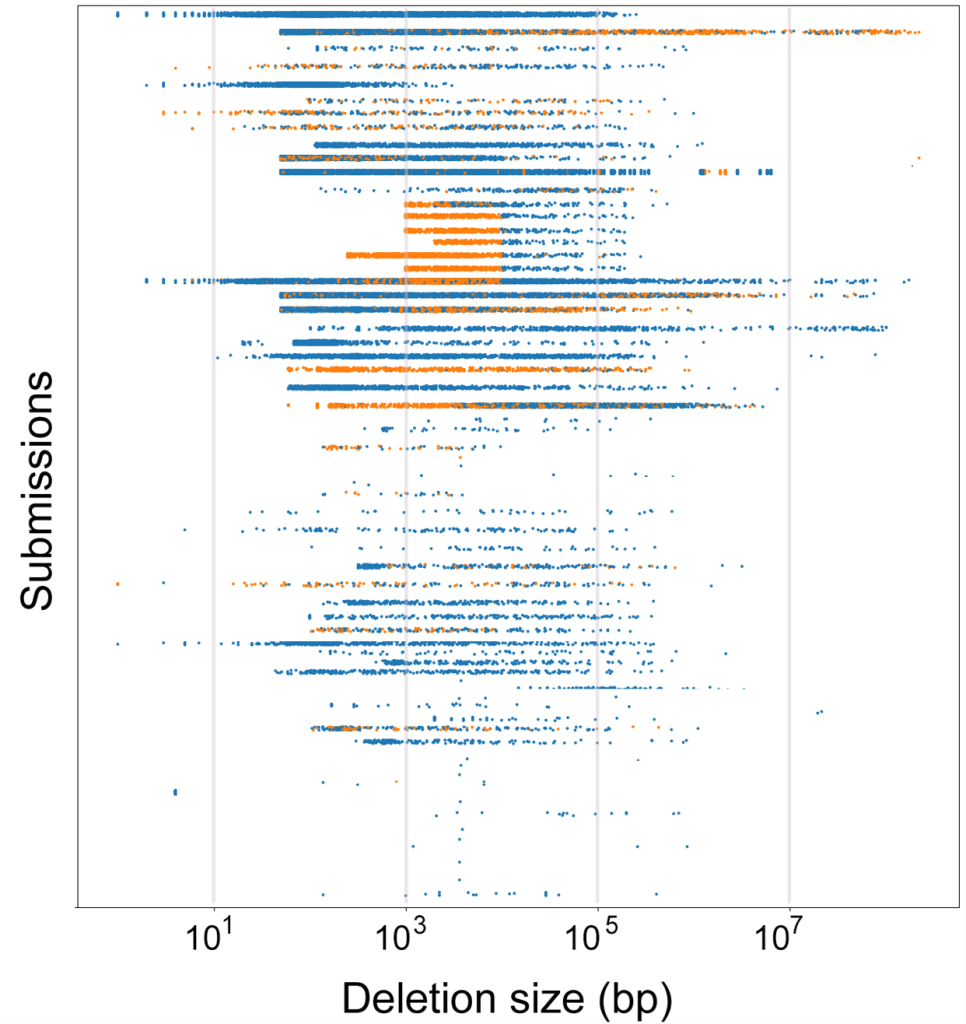
Figure 1: Sizes of copy number loss variant calls. Each dot represents a CNV loss call. Each row corresponds to a submission (only part of the
data is shown), the horizontal position of the dot corresponds to the size of the called deletion. The color of the dot represents the filtering status, with
rejected calls in orange. (Gutowska-Ding et al. 2023, doi: 10.1038/s41431-023-01482-x)
Clinical diagnostics
To improve the clinical diagnostic power of WES, it is possible to gain some extra information also from the intronic segments by adding fragmented capture through capture spiking. This has been done for example by Twist Biosciences through the creation of a CNV backbone to be combined with a standard WES capture. The value of regular spike-in grows for longer gaps not otherwise amenable to typical panel sequencing1. Also, the high uniformity and quality of the Element Biosciences sequencers’ read-out provide better precision to the breakend detection process.
This has been tested by Euformatics in a systematic validation of its full range CNV calling pipeline (exon to whole chromosome aneuploidy) combined with the Twist exome CNV backbone capture on a set of 1000 Genomes Project samples available from Coriell2 (CNVPANEL01, 43 samples). The raw sequencing data came from libraries constructed using the Twist Exome 2.0 Plus Comprehensive Exome Spike-in capture panel. The Twist spike-ins targeted polymorphic SNPs distributed in the intergenic and intronic regions. Combined with the exon targeting, this backbone of spike-ins allowed a genome-wide detection of CNVs and loss of heterozygosity (LOH) in addition to small variants with a sensitivity (recall) of 100% over 42 samples.
One sample had a uniparental disomy, which requires an adjustment in the calling process, and it is not, sensu structo, a CNV. Precision was not estimated, since the CVPANEL01 does not provide a full truth set of all the CNVs present in the samples.
Conclusion
WES together with extra spike-in provide good, even if fragmented capture coverage over the genome. A high quality sequence read-out is then sufficient for clinical diagnostic laboratories to provide exon-level CNV calling. Intra-exonic CNVs are already covered by WES without spike-in. Once CNV breakends are detected, such as with the Euformatics validated variant calling pipelines, variant analytics and reporting tool omnomicsNGS will provide the necessary support to add any essential variant, compound heterozygosity, gene, phenotype, similarity to previously identified structural variants, and other annotations to support clinical reporting.

