
Introduction
Analysing genetic variants in cancer tissue, or tumor profiling, is the cornerstone of precision oncology. It is based on the fact that mutations have a critical role in driving a specific patient’s cancer. The analysis allows clinicians to perform more accurate diagnoses, to apply better targeted therapies, and to provide better prognostics.
Tumour diagnostics based on next-generation sequencing (NGS) is increasingly expanding its scope and application within oncology with the aim of enhancing the efficacy of precision medicine for patients with cancer. The use of NGS in oncology differs from its use in constitutional genetics in being focused on treatment actionability. Action refers here to selection of drug treatments, patient enrolment in clinical trials and promotion of drug development (Mosele et al. 2024).
Sequencing of patient tumor samples generates vast volumes NGS data. Without a structured framework for interpretation and reporting, these data remain difficult to translate into meaningful clinical action. In cancer diagnostics and disease monitoring, the challenge is even greater: results must inform diagnosis, prognosis, therapeutic choice, and sometimes resistance monitoring, often under time pressure.
Somatic NGS reporting requires the integration of technical performance metrics, bioinformatic processing, biological interpretation, and clinical relevance into a concise yet comprehensive document. A well-designed report supports variant interpretation, transparent communication with oncologists and pathologists, and traceability for regulatory and quality purposes.
This article outlines the fundamental elements of a somatic NGS report, from raw data generation through variant classification and clinical interpretation. While no single universal guideline defines the exact structure of a somatic NGS report, multiple professional standards provide clear recommendations on report content. These include the AMP/ASCO/CAP standards, CAP accreditation checklists, ISO 15189 requirements, and national best-practice guidance. Together, they define the essential elements needed to ensure clarity, reproducibility, and clinical usability of somatic variant reports.
What are somatic tests in the context of cancer?
Somatic genetic tests aim to identify acquired genetic alterations that arise in cells during a person’s lifetime. These so called somatic alterations are not inherited and are typically confined to cancerous tissue.
In its simplest form, a somatic diagnostic test addresses a focused clinical question:
- Is there a molecular alteration that explains the observed phenotype?
- Does this tumour harbour an actionable mutation?
- Are there biomarkers predictive of therapy response or resistance?
For example, identifying activating EGFR mutations in lung adenocarcinoma directly informs the use of tyrosine kinase inhibitors. Similarly, detection of BRAF V600E in melanoma can determine eligibility for targeted therapy. The comprehensiveness of tumor profiling is a matter of both pragmatism and of costs: how much information is needed for supporting good actionability? An NGS test can be performed across tens, hundreds, or thousands of genes or even over a whole-genome. More data is particularly relevant in metastatic cancers of unknown primary tumour, or refractory disease where standard treatments have failed.
Somatic NGS reports serve as the primary communication channel between the molecular diagnostic laboratory and the clinical team, translating genomic observations into clinically actionable knowledge.
Somatic NGS tests are not always diagnostic
Not all somatic NGS tests are strictly diagnostic. Some are profiling or screening assays, designed to identify molecular alterations without necessarily establishing a definitive diagnosis.
Examples include:
- Broad tumor profiling panels used for therapy matching
- Liquid biopsy assays (ctDNA detection) for minimal residual disease monitoring
- Research-oriented sequencing to identify eligibility for clinical trials
- Clonal hematopoietic (CHIP) mutation screening (Reed et al. Clin Cancer Res. 2023)
While the underlying sequencing technology may be identical, the intent of testing fundamentally affects reporting. Diagnostic reports emphasize validated clinical relevance, whereas screening or profiling reports may include exploratory findings, emerging biomarkers, or variants of uncertain significance with appropriate disclaimers. Clear communication of test intent is therefore essential to avoid misinterpretation of results.
NGS as a measurement technique for somatic variation
NGS measures DNA sequences by repeatedly sampling short fragments from a heterogeneous mixture of molecules. In somatic testing, this heterogeneity is amplified by factors such as:
- Tumor purity and stromal contamination
- Subclonal populations
- Copy number variation and aneuploidy
Each sequencing read represents a probabilistic observation of a molecule drawn from this mixture. Variant allele frequency (VAF) therefore becomes a critical parameter, reflecting both biological and technical factors.
Somatic NGS reporting must address not only what variants were detected, but also with what confidence. Coverage depth, base quality, strand bias, and detection limits are essential metrics to contextualize findings and to explain the absence of expected alterations.
Tools like omnomicsQ can help laboratories systematically capture and present these quality metrics in reports. By integrating VAF, coverage statistics, and other technical parameters, omnomicsQ supports transparent reporting of variant confidence and assay limitations, ensuring that clinicians can interpret results accurately and reliably.
Tumour genomes and intratumoral complexity
Unlike the relatively stable germline genome, tumor genomes are highly dynamic. They accumulate point mutations, insertions and deletions, structural rearrangements, copy number changes, and chromosomal instability. Protein domains are functional and structural units of proteins. They are responsible for specific functions that contribute normal cellular differentiation, development, and cell interactions such as signaling cascades. Because of this essential role, many actionable variants occur in protein domains (Emerson and Chitluri. Database. 2021).
Tumours are rarely genetically uniform. Subclonal architectures mean that clinically relevant variants may be present in only a fraction of tumor cells, complicating detection and interpretation.
Certain genomic regions are inherently difficult to sequence due to GC content, repeats, or pseudogenes. In somatic reporting, these limitations must be explicitly stated, particularly when negative findings could influence clinical decisions.
The rapidly evolving knowledge landscape in oncology
Somatic variant interpretation depends heavily on continuously evolving biomedical knowledge. New therapeutic targets, resistance mechanisms, and biomarker-drug associations are reported at an unprecedented pace.
A variant classified as non-actionable today may become clinically relevant tomorrow. Conversely, early evidence may later be downgraded. Somatic NGS reports therefore represent a snapshot in time, tied to the databases, guidelines, and literature available at the moment of analysis. Many laboratories explicitly state that reinterpretation may be warranted as knowledge evolves.
What is “normal” in a tumour context?
In somatic testing, “normal” has multiple meanings. Variants are typically identified by comparison to:
- A human reference genome (e.g. GRCh38)
- Matched normal tissue from the same patient (when available)
- Population databases such as gnomAD to exclude common germline variants
Tumors, however, may carry alterations that are rare or absent in population databases but still biologically neutral. Conversely, some pathogenic driver mutations may appear at low frequency due to subclonality or technical limitations.
Distinguishing somatic from germline variants is a central challenge, particularly in tumor-only sequencing. Reports typically indicate if matched normal samples were analysed and describe the method used for germline variant filtering.
From tumour DNA to list of somatic variants
The path from tumor tissue acquisition to somatic variant list involves multiple critical steps:
- Sample acquisition and fixation (e.g. FFPE-related artifacts)
- DNA extraction and library preparation
- Sequencing and base calling
- Alignment to a reference genome
- Variant calling, filtering, and annotation
Somatic variant calling is inherently heuristic and probabilistic, particularly at low allele frequencies. Rigorous quality control is therefore essential to ensure both sensitivity and specificity. Variants are described using established standards, notably HGVS nomenclature.
Reports typically document key quality metrics such as, but not limited to:
- Read depth
- Variant allele frequency
- Coverage
- Tumor purity estimates
- Assay-specific limitations
Are all somatic variants clinically relevant?
Most detected somatic variants are passenger mutations without direct clinical consequence. Only a subset represents driver alterations with diagnostic, prognostic, or therapeutic relevance.
Two complementary concepts are central:
- Variant classification: assessing oncogenicity or biological relevance
- Variant prioritisation: ranking variants according to clinical importance in the specific tumour and patient context
A variant may be clearly oncogenic but clinically irrelevant for a given cancer type, while another may be weakly characterised but therapeutically actionable. For example, BRAF V600E is a canonical activating oncogenic mutation in both colorectal cancer and melanoma. However, its therapeutic relevance is highly tumour-type dependent. In colorectal cancer, single-agent BRAF or MEK inhibition is largely ineffective due to rapid EGFR-mediated feedback reactivation of the MAPK pathway, making BRAF V600E clinically irrelevant as a standalone target (Prahallad et al. Nature, 2012; Corcoran et al. Cancer Discovery. 2012). In contrast, in melanoma, BRAF V600E is highly actionable, with combined BRAF and MEK inhibition demonstrating substantial and durable clinical benefit, leading to regulatory approval of multiple BRAF/MEK inhibitor combinations (Long et al. New England Journal of Medicine. 2014; Dummer et al. NEJM. 2018).
Guidelines for asserting somatic variant significance
Professional guidelines support standardised somatic variant interpretation:
- AMP/ASCO/CAP guidelines for somatic variant interpretation in cancer, defining four tiers of clinical significance. Focuses on clinical utility in the specific tumor context.
- ClinGen/VICC frameworks for oncogenicity classification focus on oncogenicity and evidence curation, classify variants as oncogenic, likely oncogenic, uncertain, likely benign, or benign.Useful for biological interpretation even if therapeutic relevance is limited. Hosted on the ClinGen Cancer Variant Interpretation working group portal, which coordinates somatic variant curation: ClinGen Cancer Variant Interpretation Committee (CVI) page
These systems distinguish between strong clinical evidence, emerging evidence, unknown significance, and benign findings. Adherence to such frameworks improves consistency, transparency, and clinical trust.
From interpretation to the clinical NGS report
The recommendations outlined by Li et al. (2017) describe the essential elements that should be included in a high-quality somatic report. These include, but are not limited to:
- Patient and sample identifiers
- Test indication and assay description
- Concise summary of key findings
- Variant listings with HGVS nomenclature, transcript references, and clinical classification
- Variant allele fraction (VAF) and sequencing coverage
- Clinical interpretation, including potential diagnostic, prognostic, or therapeutic relevance
- Methodological limitations and appropriate disclaimers
Primary findings are often separated from secondary or incidental findings, each with their own interpretation. References to databases, guidelines, and software versions used should be clearly documented. Furthermore, interpretation should not be limited to positive findings Somatic reports should not focus solely on positive findings. Clinically relevant negative results should be reported in a disease-specific context, particularly for Tier I drug–cancer pairs, where the absence of a mutation directly influences treatment decisions. For example, documenting the absence of an EGFR mutation in lung cancer or a BRAF mutation in melanoma is critical for appropriate therapy selection.
Finally, while molecular laboratories provide interpretation, therapeutic decisions remain the responsibility of the treating clinician, often in the context of a multidisciplinary tumor board.
Somatic NGS report generation in omnomicsNGS
Somatic variant interpretation and report generation using omnomicsNGS consolidates interpreted molecular findings into a structured clinical document intended to support oncology decision-making. omnomicsNGS provides integrated support for both variant interpretation according to best practice guidelines and standardised report generation, ensuring that findings are classified, evidence-backed, and presented clearly for medical professionals. Each report is timestamped, indicating the date of sample receipt and report issuance, thereby defining the temporal context of the underlying biomedical knowledge and database versions used.

Figure 1: Example of report section on patient, orderer, and empiricist
The report starts with a test description, offering context about the sample and the methods employed. This is followed by an interpretation and recommendations section, where the molecular findings are summarised and placed in clinical context. When relevant, results from additional assays for example, PCR, IHC, FISH, or other non-NGS tests can also be included to provide a comprehensive overview.
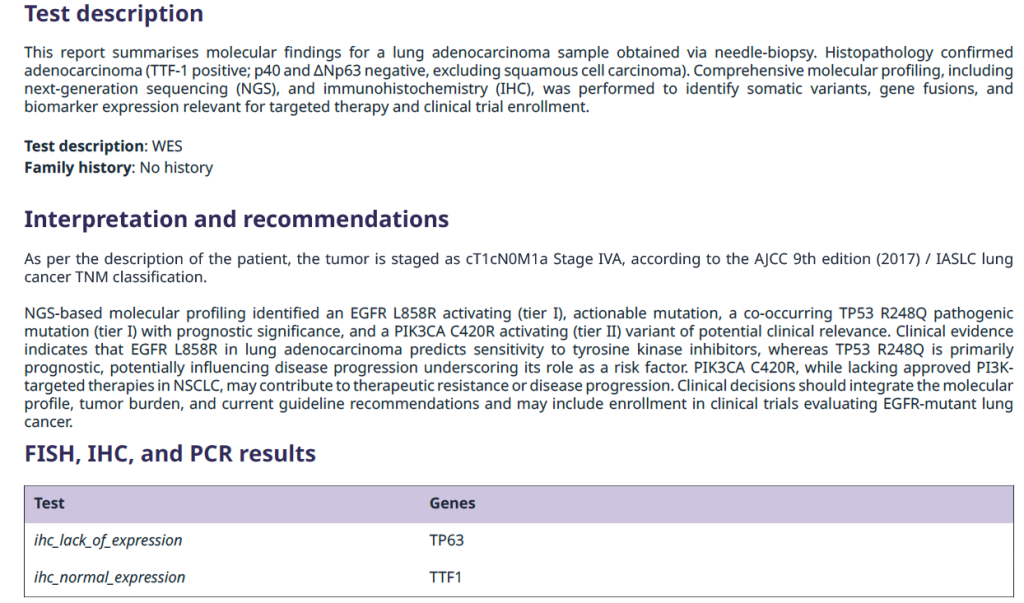
Figure 2. Example of report section on test description and interpretation and recommendations
The summary table provides a snapshot of reported Tier I and Tier II variants, with details such as gene name, HGVS notation, transcript ID, VAF %, and sequencing depth.

Figure 3. Example Summary table of a somatic report
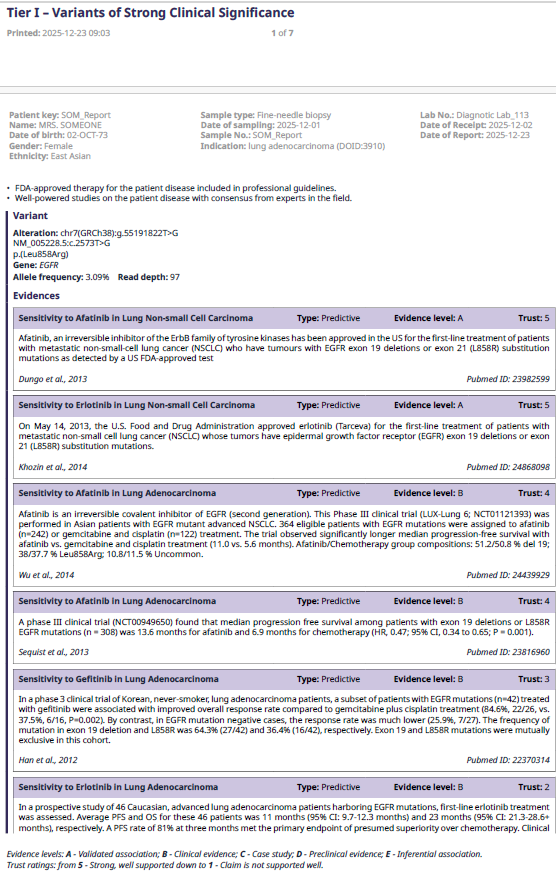
A list of evidence supporting Tier I and Tier II variants follows the summary table, sorted in descending trust level from the highest down to level 1.
A.
B.
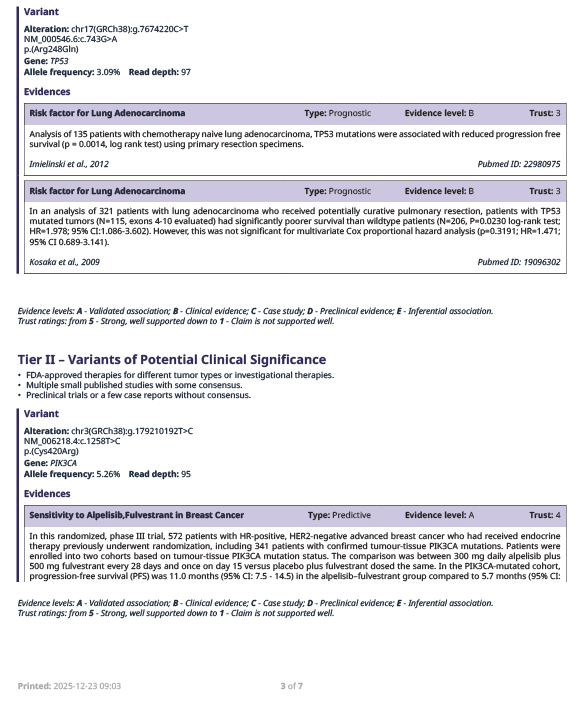
Figure 4. Example of list of evidence supporting tier I (A), and tier II (B) variants.
omnomicsNGS provides a curated list of EMA and FDA approved drugs (targeted therapies) based on user-provided ICDO-3 morphology and topography codes. Additionally, it includes ongoing clinical trials for selected diseases (DOID), which can be incorporated into the report.
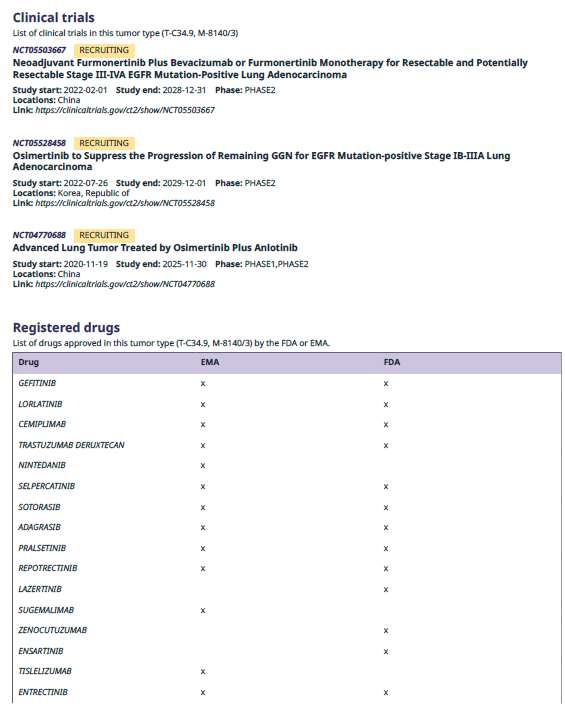
Figure 5. Example of list of ongoing clinical trials for the selected disease and EMA and FDA approved drugs for the selected ICDO-3 codes.
Relevant sample metadata for example results from MSI, HRD, and TMB analysis performed outside of omnomicsNGS can be included in the report to provide comprehensive context for each case.
Figure 6. Example of “Sample metadata” section from somatic report.
If quality control metrics are provided by omnomicsQ, they will be included in the report to ensure the reliability of the results.
Figure 7. Example of report section on quality control of NGS data
Finally, the report includes limitations of the test, variant types along with databases and their versions used for the variant analysis.

Figure 8. Example of report section on limitations of the test and applied data sources for the annotations
Reporting of structural variants and fusions follows the same format as described above, except that summary of CNV variants table contains “Protein-coding genes” and “Dosage-sensitive genes” and copy number (CN) value as indicated in input VCF file.

Figure 9. Example of a summary table for CNV variants
Conclusion
A well-structured somatic variant interpretation report is essential for translating complex NGS data into actionable clinical insights. From sample processing and sequencing to variant calling, annotation, and interpretation, each step contributes to the reliability and clarity of the final report. Incorporating standardised frameworks, quality control metrics, and evidence-backed classification ensures that reports are both clinically meaningful and compliant with professional guidelines. Tools like omnomicsQ and omnomicsNGS streamline this process, supporting consistent, transparent, and traceable reporting. Ultimately, comprehensive somatic NGS reports support oncologists and multidisciplinary teams such as molecular tumor boards to make informed decisions, optimise patient care, and adapt to the rapidly evolving landscape of cancer genomics.

